{kind=link}
por Yuta Adachi y Hiromichi Ebi

Yuta Adachi Es científico del laboratorio de Hiromichi Ebi en el Instituto de Investigación del Centro Oncológico Aichi en Nagoya, Japón. Su investigación se centra en investigar los mecanismos mediante los cuales las células tumorales mutantes KRAS escapan de inhibidores de KRAS específicos mutantes.
Hiromichi Ebi es Jefe de la División de Terapéutica Molecular del Instituto de Investigación del Centro Oncológico de Aichi y Director del Centro de Medicina de Precisión del Hospital del Centro Oncológico de Aichi. También es profesor adjunto de la División de Terapéutica Avanzada del Cáncer de la Facultad de Medicina de la Universidad de Nagoya. En su trabajo postdoctoral, el Dr. Ebi estudió estrategias para mejorar la eficacia de los agentes de orientación molecular en el laboratorio del Dr. Jeffrey Engelman en el Centro Oncológico del Hospital Basic de Massachusetts.
El reciente desarrollo de inhibidores de KRAS G12C ha llevado a un cambio de paradigma en el tratamiento de estos cánceres con mutaciones de RAS. Sin embargo, el rápido desarrollo de la resistencia a los medicamentos sugiere que es possible que éste sea un tema recurrente en la terapia dirigida. Por lo tanto, determinar los mecanismos mediante los cuales las células tumorales mutantes KRAS escapan de las agresiones inhibidoras y diseñar estrategias de combinación racionales para combatir la resistencia innata y adquirida serán fundamentales para mejorar la respuesta tumoral y los resultados de los pacientes.
La resistencia a la terapia del cáncer consiste principalmente en primaria/intrínseca y adquirida. Mientras que la resistencia primaria/intrínseca es causada por la presencia de clones resistentes a los medicamentos, la resistencia adquirida surge de la expansión selectiva de subpoblaciones preexistentes, raras y totalmente resistentes, o de la aparición de clones resistentes a los medicamentos incitados por la presión terapéutica. Estos clones resistentes pueden desarrollarse a partir de mecanismos genéticos, como mutaciones y amplificaciones, o de mecanismos no genéticos, como el recableado inducido por fármacos de la expresión genética, la señalización y las redes metabólicas que disminuyen la dependencia de una célula tumoral de la oncoproteína objetivo. En el caso de resistencia adquirida a inhibidores de KRAS G12C se han detectado mutaciones secundarias en RAS y alteraciones genómicas en la cascada de señalización MAPK 1,2. Anteriormente demostramos que la transición epitelial a mesenquimatosa (EMT) es una causa de resistencia tanto intrínseca como adquirida a los inhibidores de KRAS G12C. 3.
La resistencia adaptativa se encuentra en el medio de la intrínseca y la adquirida, en la que la amortiguación de la respuesta terapéutica es precipitada por cambios en la crimson de señalización después del tratamiento, que ocurren pocas horas después de la exposición al fármaco. 4. La resistencia adaptativa contribuye de manera importante a la insensibilidad de las células tumorales a las terapias dirigidas que bloquean la vía MAPK, ya que esta crimson está ampliamente regulada por mecanismos homeostáticos de retroalimentación negativa que afinan la producción de señales tanto en las células cancerosas como en las normales.
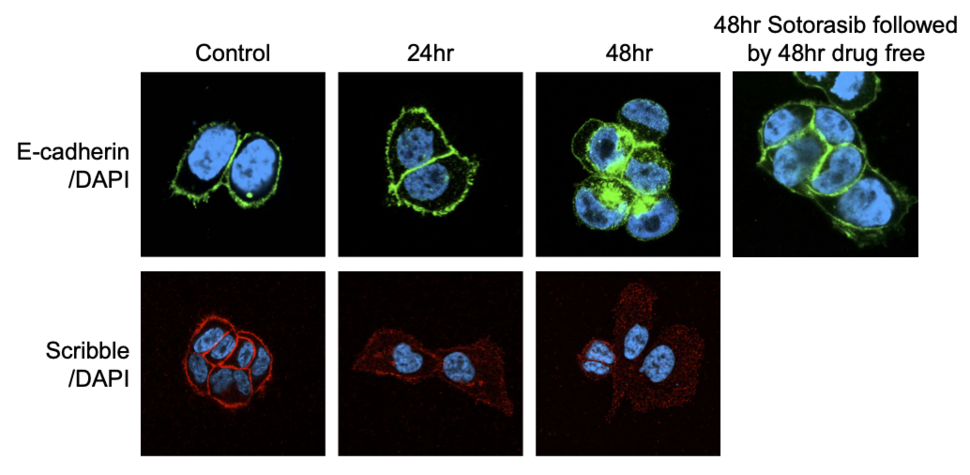
Figura 1. El inhibidor de KRAS G12C, sotorasib, induce la relocalización de las proteínas membranosas E-cadherina y Scribble.
En nuestro estudio reciente, observamos inicialmente que el proceso de EMT comienza poco después del tratamiento con inhibidores de KRAS G12C, dentro de las 48 horas. 5. La inhibición de KRAS G12C conduce a la relocalización de E-cadherina y la proteína de polaridad apical Scribble desde la membrana plasmática hasta el citosol (Figura 1). La inducción de EMT no fue causada por la regulación transcripcional de los factores de transcripción de EMT, sino por la supresión de la señalización de MAPK. Nuestro análisis preliminar sugiere que la señalización de MAPK regula positivamente los niveles de proteína de ZDHHC7, que promueve la palmitoilación de Scribble, necesaria para su tráfico de proteínas y localización en la membrana. En specific, el proceso se revirtió con la retirada de sotorasib.
Scribble regula positivamente las quinasas de la vía del hipopótamo y su regulación negativa conduce a la activación de YAP/TAZ en las células mamarias 6. El tratamiento con inhibidores de KRAS G12C también reguló negativamente la señalización de la vía del hipopótamo, lo que llevó a la translocación nuclear de las proteínas YAP y su actividad transcripcional. La combinación de sotorasib con un inhibidor de TEAD logró la reducción del tumor en vivoy estos datos están respaldados por un documento adjunto 7.
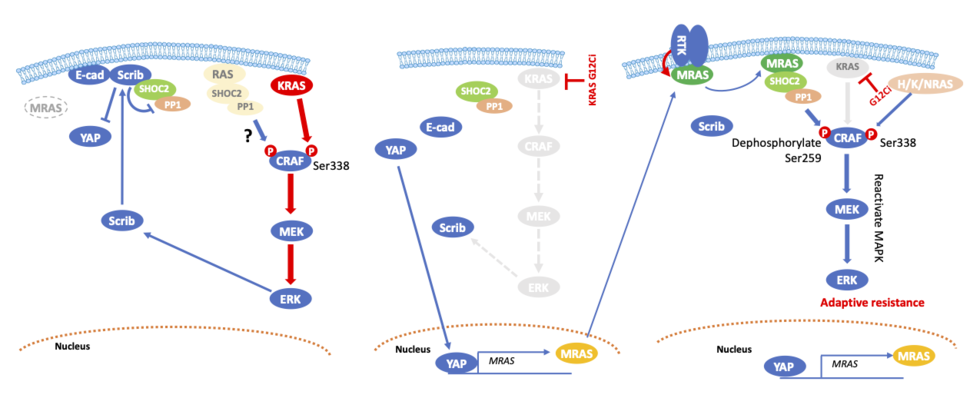
Figura 2. Diagrama esquemático de la reactivación de la señalización MAPK después de la inhibición de KRAS. izquierda. En ausencia de fármaco, el KRAS mutante activa la señalización MAPK, lo que promueve la localización en la membrana de Scribble (Scrib). Scribble forma un complejo con SHOC2/PP1c e inhibe la translocación nuclear de YAP. Medio. La inhibición de KRAS G12C regula negativamente la activación de MAPK, lo que conduce a una reducción en la localización membranosa de Scribble. Dado que Scribble no puede inhibir YAP, YAP se traslada al núcleo y transcribe sus proteínas posteriores, incluido MRAS. Bien. MRAS se traslada a la membrana donde es activado por RTK. GTP-MRAS forma un complejo con SHOC2/PP1a (el complejo MSP) que desfosforila CRAF S259. Después de la desfosforilación de CRAF, es possible que H / Ok / NRAS fosforilen CRAF S338, lo que resulta en la reactivación de la señalización de MAPK.
Además, descubrimos que MRAS es un objetivo directo de YAP. Aunque MRAS es un gen de la superfamilia RAS que es estructuralmente related a H/Ok/NRAS y comparte una secuencia de unión efectora casi idéntica en el interruptor I 8,9MRAS media su señalización a través de SHOC2, un regulador positivo de la vía RAS con, con diferencia, la afinidad de unión más fuerte entre los miembros de la familia RAS. 10. MRAS activado se une a SHOC2 y a la subunidad catalítica de PP1, reclutándolos a la membrana plasmática donde el complejo ternario desfosforila CRAF en S259, lo que resulta en la activación de CRAF. Dado que la expresión de MRAS es baja a nivel basal, Scribble interactúa con SHOC2 para inhibir la desfosforilación de CRAF S259 mediada por SHOC2. La inhibición de KRAS induce un cambio molecular de Scribble/SHOC2 a MRAS/SHOC2, lo que lleva a la reactivación de la señalización MAPK mediante la desfosforilación de CRAF S259. Dado que la reactivación por retroalimentación de la señalización de MAPK después de la supresión de KRAS G12C está mediada por receptores tirosina quinasas (RTK), inhibidores contra RTK o proteínas adaptadoras como SOS1 y SHP2 regulan negativamente MRAS-GTP (Figura 2).
Con base en nuestros hallazgos, proponemos que la relocalización de proteínas es un mecanismo de resistencia novedoso y no genético. Si bien desde hace tiempo se ha demostrado que la EMT es un mecanismo clave de resistencia, es sorprendente la rapidez con la que se induce la reorganización de las proteínas membranosas después del tratamiento con inhibidores de KRAS G12C. Además, la relocalización de estas proteínas membranosas es reversible, lo que es consistente con las nociones actuales de que la EMT no es un estado binario, sino más bien un estado de transición complicado y dinámico entre los fenotipos epitelial y mesenquimal. 11,12. Dado que la señalización MAPK está implicada en la regulación de la localización de Scribble, la relocalización de este issue se observa ampliamente después del tratamiento con agentes de direccionamiento molecular en cánceres adictos a oncogenes con señalización MAPK activada de forma aberrante. Se deben realizar más investigaciones para determinar si mecanismos adicionales pueden inducir la relocalización de proteínas después de la inhibición del KRAS mutante.
Se sabe que YAP induce independencia KRAS mutante 13,14y también está involucrado en el mecanismo de resistencia de los cánceres adictos a los oncogénes a las terapias dirigidas. 15-18. En el caso de los cánceres mutantes de KRAS G12C, el YAP activado desempeña un papel clave en el mecanismo de resistencia intrínseco, adaptativo y adquirido de estas células tumorales a los inhibidores de KRAS G12C que está asociado con el fenotipo mesenquimatoso. Sería interesante ver si la combinación de un inhibidor de KRAS G12C con un inhibidor de TEAD será tolerable y eficaz en la clínica. En specific, si bien la eliminación de MRAS previene la resistencia adaptativa a los inhibidores de KRAS G12C, la eliminación de MRAS tiene poco efecto sobre la eficacia de estos inhibidores una vez que se establece la EMT completa. Esto sugiere que YAP reescribe muchas otras redes de señalización, incluidos los procesos metabólicos y apoptóticos.
Nuestros hallazgos proporcionan detalles funcionales de una serie reciente de estudios que determinaron la estructura del complejo MSP (MRAS-SHOC2-PP1C). 19-22. El complejo MSP activo se formó solo en presencia de MRAS unido a GTP, pero su actividad fue regulada negativamente por la inhibición de RTK o proteínas adaptadoras, lo que sugiere que MRAS es el centro de activación por retroalimentación de la señalización de MAPK a través de RTK. De acuerdo con nuestros hallazgos, la eliminación de células mutantes KRAS sensibilizadas con SHOC2 a inhibidores de MEK y células mutantes EGFR a inhibidores de tirosina quinasa EGFR en NSCLC 23-25.
La naturaleza relativa farmacológica del complejo MSP justifica el desarrollo terapéutico dirigido a este complejo para evitar la reactivación por retroalimentación de la señalización MAPK. Sin embargo, primero debe establecerse el papel preciso de este complejo y su relación con otras isoformas de RAS. En el estado basal, para evitar la activación inadvertida de la vía MAPK, CRAF se fosforila en los residuos S259 y S621, lo que promueve la unión de proteínas diméricas 14-3-3 que secuestran RAF, restringiéndolo al citoplasma. Una vez que se activa MRAS, se cree que la unión de RAS-RAF desplaza a las proteínas 14-3-3 del sitio CRAF S259, lo que permite su desfosforilación por parte de PP1C en el complejo MSP. La desfosforilación de CRAF S259 por el complejo MSP precede a la activación catalítica de S338, y este parece ser el primer paso en la activación de la vía MAPK. Sin embargo, las células cancerosas mutantes KRAS positivas para marcadores epiteliales expresan niveles muy bajos de MRAS. Aunque los complejos SHOC2-H/Ok/NRAS-PP1C de menor afinidad pueden funcionar en algunas situaciones 26, el mecanismo por el cual CRAF S259 se desfosforila en células de baja expresión de MRAS no está claro. En la línea celular de cáncer de pulmón mutante NCI-H358 KRAS G12C, la desactivación de MRAS aumentó la fosforilación de ERK, presumiblemente debido a la compensación por otras isoformas de RAS 5.
Las células tumorales utilizan todos los medios posibles para evadir los fármacos dirigidos. Nuestro estudio destaca la importancia de la regulación postraduccional, como la localización de proteínas, como nuevos mecanismos de resistencia a fármacos moleculares dirigidos. YAP es una kriptonita potencial para las células cancerosas mutantes KRAS, aunque apuntar a este issue podría resultar tóxico en los pacientes. Como mediador clave de la reactivación por retroalimentación de la señalización MAPK, el complejo MSP puede ser un objetivo farmacológico atractivo para prevenir la resistencia adaptativa a los inhibidores de KRAS G12C.