{kind=link}
por Trent E. Balius y Megan Rigby

Trent E. Balius es uno de los desarrolladores del MUELLE UCSF software program, que es una herramienta computacional utilizada para predecir cómo una molécula pequeña (ligando) se une a un sitio en una proteína (u otra macromolécula). Trent dirige el equipo de Química Computacional RAS en el Iniciativa NCI RAS, donde utiliza el acoplamiento molecular para buscar nuevas terapias dirigidas a RAS. Recibió su doctorado de Universidad de Stony Brook estudiando Biología Computacional en la Departamento de Matemática Aplicada y Estadística Bajo la dirección de Robert Rizzoy realizó su formación postdoctoral en la Laboratorio Shoijeten el Departamento de Química Farmacéutica en el Universidad de California, San Francisco (UCSF).
Megan Rigby es miembro de la Grupo de inhibidores covalentes en la Iniciativa RAS como Investigador Asociado II. Actualmente está completando sus estudios de posgrado en Ciencia Biomedica en Colegio Campana.
Entrevista (transcripción editada).
megan: ¡Gracias por aceptar compartir tu trabajo con nuestros lectores, Trent! Profundicemos: ¿cómo se resolve en qué moléculas centrarse en el diseño y descubrimiento computacional de ligandos/fármacos? ¿Qué moléculas pequeñas examinas?
Trento: Creo en centrarnos en moléculas que sean accesibles, moléculas que estén disponibles para comprar y sean económicas. Tengo esta filosofía arraigada en mí gracias a mi formación en el Laboratorio Shoichet de la UCSF de que los métodos computacionales solo son útiles si puedes PROBAR la predicción.
Realizo una gran pantalla de acoplamiento en la que acople cientos de millones de moléculas pequeñas en una bolsa de proteínas y luego nuestro grupo predecirá si algunas se unirán y luego lo probaremos experimentalmente. Entonces, si desea realizar pruebas experimentales, que es la única forma en que una pantalla digital es útil, entonces debe poder comprar las moléculas (en inventory) o fabricarlas (fabricarlas bajo demanda). Las moléculas en inventory están guardadas en algún estante y usted puede solicitar una cotización para esos compuestos. Las moléculas de fabricación bajo demanda aún no existen, pero la empresa tiene bloques de construcción/piezas de moléculas que reaccionarán entre sí para formar un producto y luego le venderán ese producto. Lo más importante para la accesibilidad es que las moléculas sean económicas y puedan fabricarse.
Megan: ¿Considera que el cribado in silico es un actor importante en la búsqueda de terapias RAS y cuál ha sido su papel hasta ahora?
Trento: Creo que los métodos computacionales pueden desempeñar un papel en la orientación de RAS. En muchos de los proyectos que se están llevando a cabo actualmente en la Iniciativa RAS, como en nuestras asociaciones con compañías farmacéuticas y otros laboratorios nacionales, se está realizando mucho trabajo computacional, incluido el acoplamiento y las simulaciones de dinámica molecular. Para centrarme en un proyecto, estoy trabajando con Simanshu y su equipo. Tienen algunas estructuras cristalinas de un mutante específico de RAS con moléculas fragmentadas unidas, y he realizado una evaluación digital de esas estructuras cristalinas. estamos explorando RAE (relaciones estructura-actividad) en torno a esos hits de acoplamiento, así como con los fragmentos originales, y encontré más aglutinantes comprando análogos de esos hits de proyección virtuales (o haciéndolos trabajando con David Turner y Workforce).
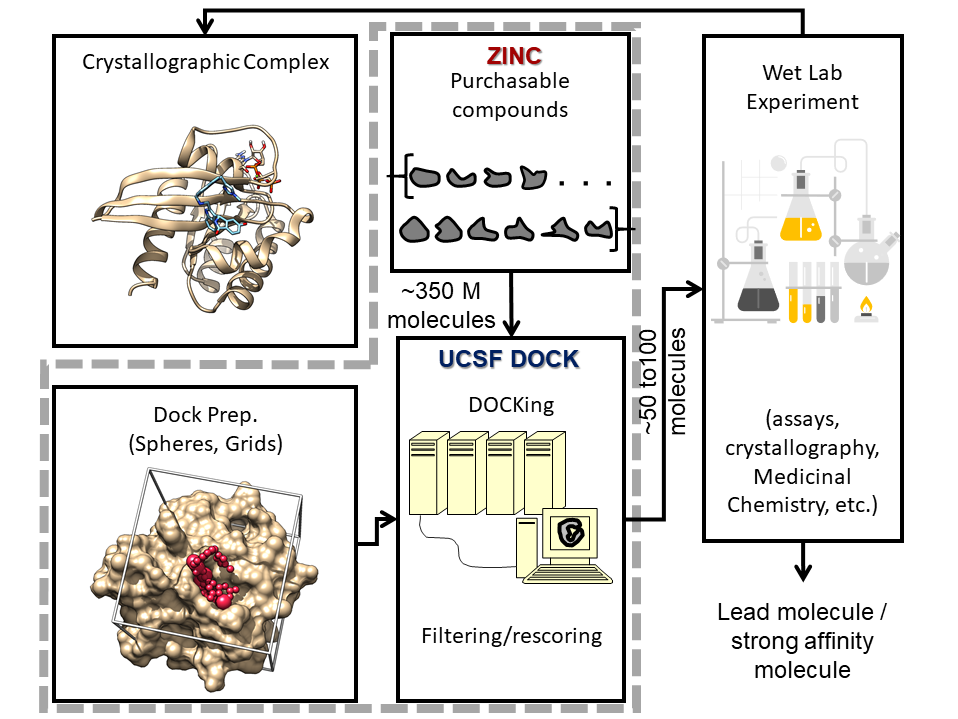
El software program UCSF DOCK se utiliza para detectar millones de compuestos adquiribles prediciendo su capacidad para unirse al RAS activo. Luego, los resultados se evalúan en el banco mediante ensayos celulares o in vitro, cristalografía y otras técnicas.
Megan: ¿Cuáles son algunos de los desafíos que ha enfrentado con RAS, en explicit, a diferencia de otras proteínas?
Trento: En primer lugar, RAS es una proteína versatile. Además, RAS tiene un cofactor, tiene GTP o PIB.. Si tiene GTP, casi siempre es un análogo en las estructuras cristalinas (un análogo no hidrolizante).
Megan: Correcto, porque el verdadero objetivo es el RAS unido a GTP: el estado oncogénico.
Trento: Correcto, el GDP (en el estado apagado) o el GTP (en el estado encendido) influyen en el estado conformacional de RAS al igual que las mutaciones asociadas al cáncer que generalmente bloquean RAS en el estado encendido.
Entonces, el cofactor es una capa de contabilidad a la que debo prestar atención cuando preparo la proteína para el acoplamiento. Tengo que asignar cargas parciales al cofactor y ahora tengo procedimientos automatizados para preparar el cofactor. Entonces, cuando hago la parametrización, también tengo que asegurarme de que los estados de protonación sean correctos, que los hidrógenos estén en los átomos correctos y que las cargas parciales sean correctas. Parece que pueden ocurrir muchos cambios conformacionales dentro de RAS y, por lo normal, al acoplarnos nos restringimos a un solo estado para la pantalla, por lo que eso puede ser un problema. Entonces, dos cosas que dificultan el modelado de RAS son la preparación del cofactor y la flexibilidad de RAS.
Megan: ¿Qué te ayuda a decidir una estructura cristalina en lugar de otra para ejecutar tu pantalla?
Trento: A menudo, las estructuras cristalinas que ya están unidas a un ligando funcionan mejor en las pantallas de acoplamiento.. Ahora mismo, muchas de las pantallas que he hecho son de estructuras cristalinas internas del equipo de Simanshu.
Megan: Parece que la detección in silico en primer lugar aumentaría significativamente la tasa de éxito. ¿Qué tan efectiva la encuentra?
Trento: Lo interesante del acoplamiento molecular como herramienta de descubrimiento de fármacos es que el acoplamiento no es muy preciso.. Si compras 100 moléculas que se acoplan bien y un experto las seleccionó de una lista de “resultados”, como máximo obtendrás 30 moléculas que se unirán, lo más possible es que obtengas 15 moléculas que se unirán.
Si estuvieras diseñando algo como un automóvil o un avión usando principios de ingeniería, entonces nuestros métodos son pésimos en comparación. Si alguien viene a mí y me pide que haga una predicción, y luego gasta miles de dólares en hacer mi predicción y no funciona, esa persona se enojará. Dado que este es el caso del acoplamiento, necesitamos múltiples tiros a portería. Por lo tanto, esperaríamos una tasa de éxito de entre el 2 y el 30% para un conjunto de moléculas seleccionadas de una pantalla de acoplamiento y probadas experimentalmente.
megan: Sin embargo, desde una perspectiva tradicional de detección de alto rendimiento, esa es una tasa de éxito bastante alta…
Trento: Pero no es tan bueno como nos gustaría. Sólo estamos probando una fracción de lo que analizamos. Entonces el acoplamiento es este filtro. Estás examinando muchas moléculas y luego tomas la parte superior de la lista para inspeccionarlas visualmente. El último paso es el filtro humano para seleccionar las moléculas.
Megan: ¿Cuánto tiempo suele llevar crear una biblioteca inicial? ¿Cuántos compuestos examina a la vez?
Trento: Utilizo la base de datos ZINC de los laboratorios Shoichet e Irwin de la UCSF. John Irwin y Brian Shoichet crearon la base de datos ZINC a mediados 2000. ZINC significa «ZINC no es comercial» y consta de moléculas extraídas de los catálogos de los proveedores. Hay un Página internet para la base de datos ZINC donde se pueden descargar moléculas preparadas para análisis computacional. ZINC22 será una renovación y tendrá muchas más moléculas. El espacio químico accesible ha crecido enormemente gracias a estos catálogos de fabricación bajo demanda y también lo ha hecho ZINC. Es un buen problema tenerlo, pero resulta un poco intimidante. Las preguntas son ¿Cuán grande es muy grande? ¿Y cómo podemos seguir el ritmo?
Como referencia, si acople compuestos similares al plomo (una cierta porción de espacio químico con un peso molecular y un rango de hidrofobicidad), que son aproximadamente 330 millones de moléculas con mi copia de ZINC.[1][2], paso aproximadamente 1 segundo por molécula y estoy usando alrededor de 1000 núcleos de CPU. Luego, se necesitan entre 3 y medio y cuatro días. Ahora, ampliemos la escala. Si desconecto 500 millones, serán 5 días, y si desconecto mil millones, serán 10 días. Si desconecto diez mil millones, son 100 días. No podemos darnos el lujo de pasar tanto tiempo en una pantalla. La única manera de que eso sea factible es si hiciéramos el software program más rápido para que podamos obtener los resultados en un tiempo más razonable.
Megan: Entonces, ¿habrá un límite para la cantidad de compuestos en una pantalla o hay cosas que la gente está haciendo para que el proceso sea más eficiente?
Trento: Una cosa que otras personas están haciendo es utilizar el aprendizaje automático para entrenar con un conjunto más pequeño de moléculas., entonces harán una pantalla y luego entrenarán un modelo de aprendizaje automático en esa pantalla más pequeña, y luego lo extrapolarán a una pantalla más grande, y luego acoplarán los que obtengan una buena puntuación. Entonces crean un método de pseudoacoplamiento[1][2]. También vi una charla en la que hicieron una pantalla dispersa y luego se centraron en diferentes secciones de la base de datos, por lo que hicieron una pantalla dinámica en la que examinaron un conjunto aleatorio y luego, basándose en eso, ampliaron una región para examinar, por lo que no examinarán toda la base de datos, se centrarán en la parte que parece interesante.
Megan: ¿Cree que la IA impulsará mejoras en el proceso de acoplamiento en el futuro?
Trento: Sí, creo que hay muchos creyentes en la IA. El El desafío con el aprendizaje automático y la IA es que necesitas muchos datos para entrenarlo. Y, a menudo, en el descubrimiento de fármacos, los datos son escasos.
El descubrimiento de fármacos es difícil por varias razones. La easy unión es difícil de entender, pero luego están todos estos otros factores de confusión, como la agregación, que es cuando las moléculas pequeñas se agregan y generan problemas. Y también tenemos estas cosas raras, como «acantilados de afinidad» donde un pequeño cambio en la estructura química del ligando puede abolir la unión. Otro problema es que la literatura científica está plagada de artefactos. Entonces, la gente afirmará que estas moléculas se unen, pero en realidad no se unen como la gente cree que lo hacen. Por ejemplo, muchas moléculas se unen de forma no específica y tienen un efecto celular, pero no actúan a través del mecanismo que la gente cree.
Por lo tanto, para que el aprendizaje automático sea una herramienta eficaz para el descubrimiento de ligandos, se necesitan datos limpios y muchos datos. La otra preocupación sobre el aprendizaje profundo (las redes neuronales) es que no se sabe lo que sucede bajo el capó.
El aprendizaje automático puede mejorar las cosas, pero no creo que vaya a resolver todos nuestros problemas.
Megan: Bien, parece que hay mucho entusiasmo desmedido en torno a la IA en este momento. Entonces, para ser claros, si dos personas entrenaran un algoritmo de aprendizaje automático en dos conjuntos de datos diferentes, ¿podrían obtener dos resultados completamente diferentes?
Trento: Sí, Las limitaciones del aprendizaje automático es que tu conjunto de entrenamiento controlará lo que encontrarás. Así que déjenme decirles mi parcialidad: estoy muy a favor de los modelos basados en la física. Creo que estos métodos de aprendizaje automático de IA son realmente útiles y útiles, pero se basan en el conocimiento, no en la física y, como usted cube, se trata de los datos que incluye. Sin embargo, hay personas que también utilizan la IA para entrenar modelos físicos y lograr que la física sea correcta.
Creo que esta concept de utilizar diferentes funciones de puntuación para diferentes proteínas no es satisfactoria. Es mucho más satisfactorio pensar que estamos entendiendo bien la física. No me malinterpretes, el aprendizaje automático es muy útil.
Megan: También puedo entender lo que quieres decir con las limitaciones de los enfoques basados en el conocimiento; como con la estructura de las proteínas, todavía hay muchas incógnitas con respecto a los estados conformacionales entre los mutantes RAS y los estados GDP/GTP…
Trento: Sí, solo les daré un ejemplo de un método de aprendizaje automático que ha cambiado el campo que es pliegue alfa. El pliegue alfa es responsable de un salto en nuestra capacidad para predecir la estructura de las proteínas. De hecho, algunas personas dicen que el problema del plegamiento de proteínas se ha resuelto. Puedes darle una secuencia alfa veces y predecirá una estructura. Pero como acabas de decir, para RAS, es una proteína muy versatile. Existen estados de bucle que cambian de conformación. Hay un sesgo hacia diferentes estados: ya sea el PIB o el GTP, la conformación será diferente. Y luego tienes la unión de efectores y cosas así. Aunque el pliegue alfa puede predecir el pliegue de una proteína, no muestra qué se une a él. Curiosamente, deja el sitio de unión de nucleótidos allí sin nucleótidos. He estado usando alfa veces para modelar proteínas y complejos de proteínas y es un ENORME avance.
Megan: ¿Tienes alguna concept closing?
Trento: El El futuro es brillante para los métodos computacionales que ayudan al descubrimiento de fármacos, con mejoras que se producen desde diferentes direcciones. Crecimiento en el espacio químico, avances en los métodos (incluida la IA), mejoras en el {hardware} (como más espacio en disco y GPU). Sí, Creo que el futuro es brillante para el descubrimiento computacional de fármacos (ligandos).