{kind=link}
de Olesja Popow y Kevin Haigis

Olesja Popow Es investigador postdoctoral en el laboratorio de Kevin Haigis en el Instituto del Cáncer Dana-Farber y está coasesorado por Steven Gygi en la Facultad de Medicina de Harvard. Su investigación se centra en definir y comprender la actividad oncogénica de KRas específica de tejido. Para ello emplea una combinación de proteómica, biología molecular y modelos de ratón. Su proyecto es parte del SPECIFICANCER Most cancers Grand Problem, financiado por Most cancers Analysis UK y la Mark Basis for Most cancers Analysis.
Kevin Haigis es profesor asociado de medicina en la Facultad de Medicina de Harvard y director científico del Instituto del Cáncer Dana-Farber. Durante su formación de posgrado en la Universidad de Wisconsin-Madison, el Dr. Haigis estudió la genética somática del inicio del tumor intestinal bajo la dirección del Dr. William Dove. En su trabajo postdoctoral, el Dr. Haigis estudió las propiedades oncogénicas de las GTPasas RAS en el laboratorio del Dr. Tyler Jacks en el Centro Tecnológico para la Investigación del Cáncer del Instituto de Tecnología de Massachusetts (ahora Instituto David H. Koch para la Investigación Integrativa del Cáncer).
Entre todos los protooncogenes conocidos (genes que, cuando se activan mediante mutaciones o amplificaciones, pueden transformar células normales en células tumorales), KRAS es uno de los que muta con más frecuencia en los cánceres humanos.1,2. Esto se alinea bien con nuestro conocimiento cada vez mayor sobre su papel central como impulsor de las vías de señalización de células protumorales.3.
Sin embargo, los datos tanto de humanos como de ratones sugieren que KRas, de hecho, no es un oncogén «common» y que la capacidad de KRas mutante para impulsar el inicio y/o la progresión del tumor se limita a un subconjunto de tejidos. De hecho, cada estudio que aplicó modelos de ratón para impulsar la expresión oncogénica de KRas a niveles endógenos en múltiples tejidos en el animal adulto observó respuestas notables específicas de tejido; A pesar de la appreciable variación en los modelos utilizados, se encontró comúnmente que el pulmón, el tracto digestivo, el sistema hematopoyético y la mucosa escamosa eran permisivos a los efectos oncogénicos de los KRas mutantes. Por el contrario, ninguno de los estudios previos detectó cambios histológicos en tejidos como el hígado, el riñón, el músculo o el cerebro.4–9. También pudimos reproducir este resultado nosotros mismos (ver Figura 1).
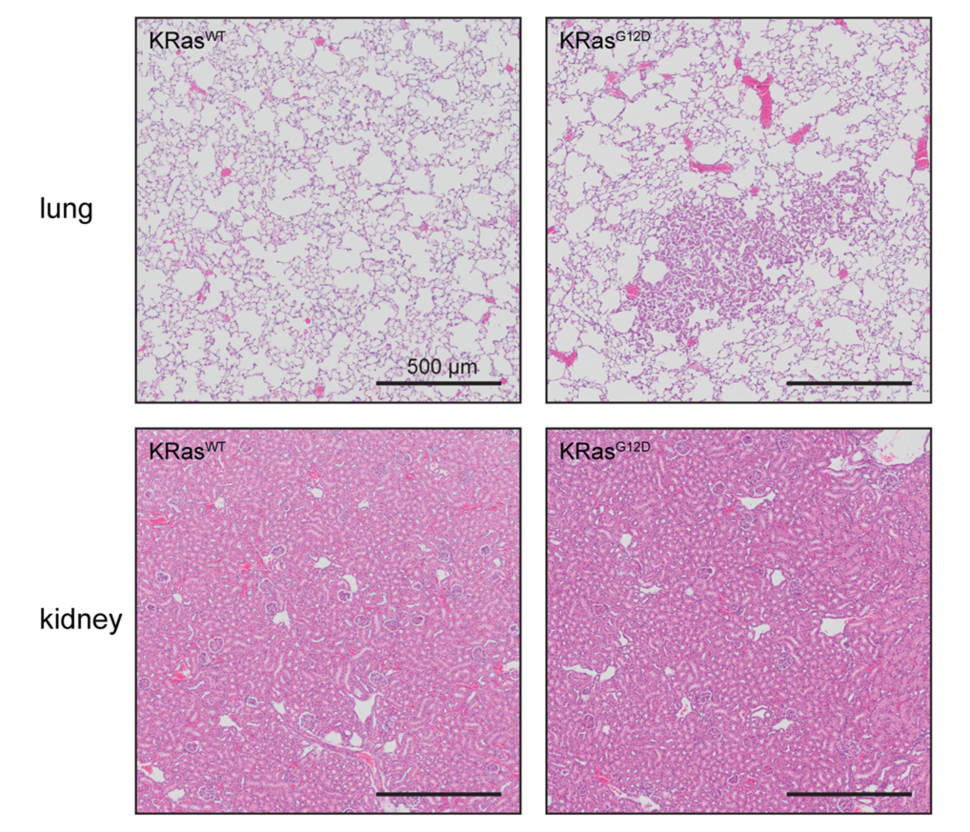
Figura 1. Secciones de pulmón y riñón teñidas con hematoxilina-eosina de ratones que expresan la proteína KRas de tipo salvaje (KRasWT) u oncogénica (KRasG12D). La introducción de KRas mutantes conduce al desarrollo de lesiones hiperplásicas en el pulmón, mientras que el riñón parece histológicamente regular.
Aunque ha sido reconocida durante más de 20 años, la actividad oncogénica específica de tejido de KRas ha permanecido en gran medida inexplorada. El patrón de permisividad del tejido KRas observado en ratones refleja en gran medida la frecuencia de mutación KRAS específica del tipo de cáncer observada en pacientes, lo que indica la aplicabilidad de modelos de ratón para el estudio de este fenómeno.2. En explicit, KRas se expresa de forma ubicua y su abundancia de proteínas no se correlaciona con la permisividad de un tejido determinado.10,11. Sin embargo, los estudios existentes sobre la función de Ras en tejidos permisivos, así como los conocimientos sobre la biología básica de Ras, sugieren posibles mecanismos subyacentes.
Hablando de manera simplista, la activación de Ras requiere su asociación con una membrana y un issue de intercambio de nucleótidos de guanina (GEF), que promueve la conversión de la forma inactiva unida al GDP a la forma activa unida al GTP. Ras unido a GTP puede posteriormente interactuar y activar proteínas efectoras, desencadenando la señalización a través de vías posteriores. La terminación de la señal se produce mediante la acción de las proteínas activadoras de GTPasa (GAP), que convierten a Ras nuevamente en su estado inactivo unido a GDP.3. No sorprende que un examen más detenido revele un panorama sumamente complejo.
La interacción de KRas con las membranas se rige principalmente por su extremo C y las modificaciones postraduccionales (PTM) ubicadas dentro de él. Los nanoclusters de KRas en la membrana plasmática ensamblan una combinación distinta de lípidos, que a su vez determinan los efectores que pueden reclutarse y activarse. Las composiciones de membrana que son desfavorables para la formación de nanoclusters pueden anular la señalización de KRas12. Del mismo modo, los cambios en los PTM C-terminales de KRas afectan la composición de lípidos de los nanoclusters y, a su vez, la salida de señalización aguas abajo.13. Es concebible que el perfil lipídico específico del tejido y el patrón de expresión de las enzimas modificadoras de Ras puedan influir sustancialmente en la formación de nanoclusters de KRas y la activación del efector.14,15.
La membrana plasmática (PM) es el sitio principal de activación de Ras, pero la señalización mediada por Ras también puede ocurrir desde las endomembranas, más notablemente el Golgi, con marcadas diferencias en la cinética y la emisión de señales.16-18. Estos conocimientos provienen principalmente de experimentos con N- y HRas, que comparten un alto grado de homología de secuencia con KRas fuera de su región hipervariable ubicada en el extremo C-terminal. No obstante, NRas, HRas y la isoforma de empalme KRas, KRas4A, utilizan la misma ruta de procesamiento postraduccional, incluida la palmitoilación C-terminal en el Golgi.dieciséis. Por tanto, es posible que la señalización mediada por KRas4A tenga lugar en el Golgi además del PM. Se cree que los RasGEF, los andamios efectores y otros coactivadores con localizaciones subcelulares restringidas impulsan las distintas dinámicas y resultados entre PM y la señalización de Ras localizada en la endomembrana.18. Si estas proteínas controlan el equilibrio entre la señalización de KRas desde diferentes compartimentos subcelulares, la variedad en sus niveles de expresión podría tener efectos profundos en los resultados de la activación de KRas entre tejidos.
La compartimentación también ocurre aguas abajo de las vías de señalización activadas por Ras. Probablemente esto se entienda mejor en el contexto de las quinasas MEK1/2 y ERK1/2, que se activan secuencialmente aguas abajo de las quinasas RAF. La unión de ERK (sola o en complejo con MEK) por estructuras subcelulares específicas del compartimento controla su capacidad de translocarse al núcleo y, por lo tanto, puede cambiar la especificidad del sustrato de ERK de sustratos nucleares a citoplasmáticos.dieciséis. Por lo tanto, las diferencias específicas de tejido en el perfil de expresión de estas proteínas de andamiaje podrían sesgar la actividad de la quinasa hacia un conjunto distinto de sustratos, dando forma así a la respuesta de un tejido a la activación de ERK por parte de KRas. En apoyo de esta hipótesis, Parikh et al. Se observó una localización nuclear diferencial de ERK fosforilada activa entre tejidos en ratones que expresan el mutante oncogénico KRasG12D.9.
Los KRas pueden interactuar y emitir señales a través de una variedad de efectores, muchos de los cuales aún no se han estudiado y es posible que solo desempeñen funciones importantes en determinadas condiciones. De manera comparable, KRas puede ser activado e inactivado por numerosos GEF y GAP, pero se desconoce cómo contribuyen a la señalización diferencial de KRas.
A pesar de la falta de comprensión detallada, está claro que una multitud de componentes de la vía Ras tienen el potencial de influir en si se activan las señales de KRas en una célula, dónde y durante cuánto tiempo. La presencia o ausencia de elementos posteriores puede dar forma aún más a la salida de señalización. Por lo tanto, planteamos la hipótesis de que es el equilibrio entre los perfiles de expresión colectiva de esta purple subyacente lo que da forma a los resultados divergentes de la activación de KRas en contextos, por ejemplo, en diferentes tejidos. De hecho, los cambios de señalización inducidos por mutantes oncogénicos de KRas, medidos mediante fosfoproteómica, difieren significativamente entre el colon y el páncreas.19,20. En casos extremos, la purple preexistente puede no soportar la señalización oncogénica de KRas, y esto podría explicar la aparente falta de respuesta en tejidos que no son permisivos a los efectos de KRas mutantes (ver también 21).
Esta hipótesis implica además que los cambios en la purple subyacente afectarán la permisividad de KRas y esto podría provocar cambios en el tropismo del tejido durante el desarrollo o la inflamación. En teoría, una variedad de estados de la purple podrían bloquear o permitir la señalización oncogénica de KRas. En consecuencia, esto plantea la cuestión de si todos los tejidos no permisivos (o, de hecho, también los permisivos) son creados iguales. Es decir, los mismos nodos dentro de la purple definen la permisividad en todos los tejidos. Es poco possible que un solo issue sea el único determinante, ya que representaría un objetivo ultimate para las comutaciones y se esperaría que fuera seleccionado en tumores de pacientes. De hecho, la inactivación de la línea germinal de p53 (el supresor de tumores mutado con mayor frecuencia en los cánceres humanos) en un ratón que expresa un alelo KrasG12D latente aumentó principalmente la malignidad del tumor, pero no alteró sustancialmente la variedad de tejidos en los que se desarrollaron las lesiones. Una excepción fueron los tumores originados en los vasos sanguíneos (hemangiosarcomas) y en el tejido conectivo (fibrosarcomas), que sólo se observaron en ratones con doble mutación p53/Kras.4.
Algunas mutaciones, solas o en combinación con la sobreexpresión, pueden aumentar la actividad de KRas más allá de un punto óptimo que sustenta la proliferación, lo que resulta en senescencia celular y apoptosis.22,23. Esto por Li et al. El denominado «punto óptimo» probablemente varía entre los tejidos y, de manera concordante, los diferentes tipos de cáncer seleccionan distintas (co)mutaciones de KRAS.23,24. Sin embargo, Le Roux et al. pero no pudo detectar ningún cambio histológico en la mayoría de los tejidos.25.
Dada la frecuencia y las dificultades asociadas con la localización de mutaciones de KRAS en cánceres humanos, existe una necesidad urgente de nuevos enfoques terapéuticos. Aunque inicialmente puede parecer contradictorio, sostenemos que los conocimientos adquiridos al estudiar los efectos de la expresión oncogénica de KRas en tejidos no permisivos serán muy valiosos para lograr este objetivo. Comprender los principios que subyacen a la permisividad de los KRas oncogénicos nos permitirá en última instancia aprovechar en vías terapéuticas las estrategias mediante las cuales algunos tejidos pueden evidentemente bloquear los efectos perjudiciales de los KRas mutantes.